癌变
癌变(英語:)通常用來形容正常细胞转变成癌细胞的致癌过程,是细胞DNA受损突变后发生在细胞及基因级别上的、从而导致细胞不受机体控制恶性增殖的一系列改变,最终引起恶性肿瘤的形成。癌变是个复杂、受到多种因素控制的多阶段演变过程,它也是个可逆转的细胞转化过程。细胞内基因的突变和肿瘤的发生与遗传,环境等多种因素有关[1][2]。
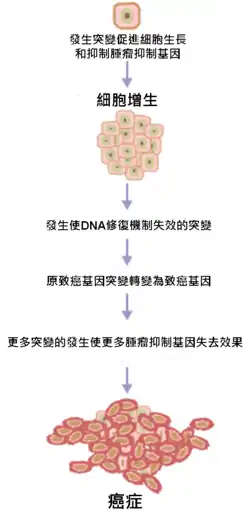
过程
正常细胞的DNA中含有原致癌基因和肿瘤抑制基因两种调控细胞周期的基因:原致癌基因有促进细胞生长、有丝分裂和分化等正常过程的作用;肿瘤抑制基因则抑制细胞过度生长、增殖从而预防肿瘤的形成。一般在细胞DNA受损时,肿瘤抑制基因会停止细胞继续分裂来修复DNA,并在不可修复时使受损细胞进行细胞凋亡。而在致癌物质的作用下,这两种基因发生突变使原致癌基因被激活成为癌基因、肿瘤抑制基因失效,导致该细胞的生长、分裂失去控制,变成持续生长和增殖的癌细胞。
因素
分子生物學
癌变意味著一連串由DNA受損而引發細胞分裂速率失控,導致癌症發生的過程。癌症是基因引起的疾病,當調控細胞生長的基因發生突變或損壞時,使得細胞失去控制,持續的生長及分裂而產生腫瘤[5]。大部分人體內的細胞是不會持續分裂生長的,除非遭遇受損,例如肝細胞、心肌細胞。但是像是由上皮細胞組成的組織,包含腸黏膜、皮膚等,均需借由複製生長來持續更新以保持功能正常。而持續的更新這些上皮細胞構成的組織是有其必要性存在的,這樣的作用可保護人體本身保持正常功能。因為上皮細胞所處的環境常接觸到外界物質或機械力的損傷,如果不能夠將受損細胞更新,必定會影響到其功能。但是具有持續生長能力的細胞,對癌症的產生就是最好的環境,對於要將其轉變成癌細胞就會簡單的多。這也是為何所有常見的癌症,多數源自於上皮細胞的原因。調控細胞生長主要有兩大類基因,原致癌基因主要是一些參與促進細胞成長、進行有絲分裂的基因。肿瘤抑制基因,則是負責抑制細胞生長或是調控細胞分裂進行。一般而言,突變需要發生在調控細胞生長的重要基因上,才有機會使一個正常細胞轉化成癌細胞。[6][7]
原致癌基因透過不同途徑促使細胞成長。有些原致癌基因可調控產生刺激細胞有絲分裂的激素,(又稱作荷爾蒙,是一種在細胞間傳遞控制訊息的「化學信號」),受到激素刺激的細胞或組織的反應則受其細胞內的訊息傳遞路徑決定。有的原致癌基因也負責組成細胞訊息傳遞系統或訊息受器,借由基因表現量的調控進而控制訊息傳遞系統對激素的敏感程度。此外分裂原、轉錄與蛋白質合成都常見原致癌基因的參與[7]。原致癌基因的突變可能影響基因表現或是功能,導致下游蛋白質的表現或活性改變。這樣的情形發生時,原致癌基因就轉變成為致癌基因,帶有致癌基因的細胞則有更高的機率發生異常。因為原致癌基因參與調控的細胞的功能十分廣泛,包括細胞生長、修復和維持體內平衡,所以我們也無法將其從染色體中去除來避免癌症發生[8]。
腫瘤抑制基因產生的蛋白質主要的功能在於抑制細胞成長、調控有絲分裂和細胞複製的過程[9]。通常是當細胞受到環境改變或DNA受損時而表現出來的轉錄因子。當細胞偵測到發生DNA損傷時會活化細胞內的修補訊息傳遞途徑,借此促使調控細胞分裂的腫瘤抑制基因表現使細胞分裂暫停,以進行修復損壞的DNA,而DNA損傷才不會傳遞到子細胞。最有名的腫瘤抑制基因為p53蛋白質,其本身是一個轉錄因子,可被細胞受到壓力後所產生的訊號所活化。例如,缺氧或是受到紫外線照射。在將近一半的癌症中,可發現p53功能缺失或是表現量異常。目前較確切的兩個作用分別是在細胞核中作為轉錄因子,以及在細胞質中參與調控細胞週期、分裂和凋亡[10]。對於p53在細胞訊息調控以及細胞生長、凋亡的功能已經有著數量極多的研究報告[11][12][13]。許多基因剔除的研究也指出p53對於細胞的重要性[14][15],所以p53在癌症的發展中必定扮演關鍵的角色,可說是研究癌症極重要的一個蛋白質[16]。
瓦氏效應是指為了維持腫瘤快速成長所需的能量,讓細胞偏向進行醣解作用作為能量來源。從有氧代謝轉換成醣解作用的過程則受到p53調控。SCO2(Synthesis of Cytochrome c Oxidase 2)被認為是瓦氏作用的主要因子,其能在粒線體內調控細胞色素c氧化酶複合體,p53則控制SCO2基因的表現,這條路徑提供p53如何參與瓦氏作用機制的解釋[17]。
然而,突變可能損及活化腫瘤抑制基因的機制或是腫瘤抑制基因本身,使得腫瘤抑制基因「被關掉」,造成修復損傷DNA的機制停止。於是DNA損傷就持續累積,而不可避免地導致癌症發生。
由於原致癌基因轉變為致癌基因的突變,會受到有絲分裂過程中的檢查機制和腫瘤抑制基因抑制。因此一般來說,癌症的發生需要兩個前提,第一是原致癌基因的突變;第二則是腫瘤抑制基因的突變。此種過程稱為努德森假說。當一個腫瘤抑制基因發生一個突變之後,由於仍有許多具有相似功能的「後備」基因可做替補,所以並不足以引發癌症。只有在原致癌基因改變成致癌基因或是損壞、不活化的腫瘤抑制基因的數量達到足夠讓促使細胞成長的信號超過正常調控細胞的訊息,細胞才會進入失去控制的快速生長。此外隨著年紀增長,突變的機率增加,細胞失去控制的機會也會增加。
但是由於DNA的損壞可形成反饋現象,努德森所提出的模型也受到質疑。有研究發現在某些腫瘤抑制基因裡,只要有一個等位基因失去作用就足以導致腫瘤產生。這種現象稱為單一等位基因不足性,也經過一定數量的實驗方法證實其存在。單一等位基因的不足性引發腫瘤生成相較於努德森假說需要較長的時間[18]。
通常致癌基因是顯性的,代表獲得功能的突變(gain-of-function mutations),發生突變的腫瘤抑制基因是隱性的,代表失去功能的突變(loss-of-function mutations)。每個細胞中同一個基因都有兩個拷貝分別來自父親和母親。一般說來,只要原致癌基因的兩個拷貝之中的一個發生突變,就足以產生得到功能的突變使其轉變成致癌基因。而要使腫瘤抑制基因發生失去功能的突變,則需要兩個拷貝都被破壞。然而雖然有時腫瘤抑制基因僅有一個拷貝突變,但此突變的拷貝會使正常的拷貝不能作用,使得基因仍然失去作用,這種現象稱作顯性負面效應(dominant negative effect),在許多p53的突變中可觀察到此現象。
致癌基因的得到功能的突變和腫瘤抑制基因的失去功能的突變,常常會使用汽車的油門與煞車來做比喻。當細胞生長是一台車子時,致癌基因就等同於油門,而腫瘤抑制基因就是這輛車的煞車,當煞車並未失效時,即使踩下油門,仍可用煞車使車停下。但如果是煞車失效時,即使輕踩油門,車子仍會前進。大致說來,致癌基因與腫瘤抑制基因的定義通常來自於一個基因對細胞生長的影響。致癌基因扮演促進細胞生長繁殖的角色,腫瘤抑制基因則抑制細胞週期進行。但是在調控細胞生長中有許許多多的因子參與,要精確的定義一個基因究竟是致癌基因或腫瘤抑制基因則需要許多不同面向的實驗結果來加以證實[19]。
腫瘤抑制基因的突變也可遺傳到下一代的基因體中,使後代增加癌症發生的機會。有許多的家族因為遺傳到帶有突變的腫瘤抑制基因而對於某些癌症有較高的發生機率。通常是來自父或母其中之一的基因拷貝帶有瑕疵。由於腫瘤抑制基因的突變通常是隱性的失去功能的突變,含有一份突變拷貝的基因,雖然能借着另一份正常拷貝來維持基因功能,但是具有瑕疵的基因就變得較正常基因更容易產生問題。例如,帶有突變p53異型合子的人就經常是李-佛美尼症候群的患者,而有视网膜母细胞瘤基因(Rb)突變的異型合子的人則是視網膜母細胞瘤的高風險群[20][21]。類似的狀況也發生在APC基因,這是與大腸直腸癌發生有關的腫瘤抑制基因,而BRCA1和BRCA2基因的突變則和乳癌相關。
癌症的根源,可以歸結於DNA突變的累積。而突變的累積則導致促進細胞生長的蛋白質大量表現,並且破壞腫瘤抑制基因的功能,使得細胞週期控制失常。引起突變的物質被稱為致變原,其中可導致癌症的致變原,則稱為致癌物質。不同致癌物質可引發不同的癌症。例如抽菸吸入的化學物質可導致肺癌;長期曝露於紫外線照射可導致黑色素瘤以及其他皮膚腫瘤的產生;吸入石棉纖維可導致間皮瘤等。例如慢性發炎也是誘發癌症的原因之一[22],由於持續發炎引起細胞的生長調控的改變,導致細胞轉化。此外廣義而言,細胞內產生的自由基由於可造成基因突變,也可算是一種致變原,慢性發炎所產生的嗜中性顆粒白血球就會分泌自由基造成DNA突變。還有染色體易位,例如费城染色体就是一種染色體之間互相交換的特殊突變。
雖然有許多致變原就是致癌物質,但是有些致癌物質卻不是致變原。例如酒精和雌激素,它們能直接促進細胞加速進行有絲分裂而增加癌症發生的機會。加快速度的有絲分裂在進行DNA複製的階段時,負責修理DNA的酵素只能使用較少的時間去修補損壞的DNA,因此也增加DNA複製出錯的可能性。在有絲分裂期間所發生的錯誤,則可能導致接受基因的子細胞染色體數目異常而引起癌症。
此外許多癌症起源於病毒感染。特別是在動物中,例如鳥類。由病毒引起的人類癌症大約佔所有人類癌症的15%。與癌症有關的病毒主要有人類乳突病毒[23]、乙型肝炎病毒、人類疱疹病毒第四型和人類嗜T細胞病毒()。實驗結果和流行病學數據顯示在所有引起癌症的危險因子中,病毒排名第二,僅次於菸草[24]。病毒引發腫瘤的方式可以分為急性轉化或慢性轉化兩種。可造成急性轉化的病毒中帶有病毒癌基因,是非常活躍的致癌基因。當被感染的細胞表現病毒致癌基因時,就會使細胞轉化。相反的,進行慢性轉化的病毒通常要將其染色體插入宿主的基因中,而這樣的過程也是逆轉錄病毒的特性。當病毒的基因插入到原致癌基因附近時,借由病毒基因帶有的啟動子或者其他調控轉譯的機制,讓原致癌基因大量表現使細胞生長失去控制。因為病毒基因是以隨機的方式插入到宿主的基因中,如果插入的地方恰好沒有原致癌基因存在,對於細胞的生長就不會有太大影響。相對於急性轉化的病毒本身即攜帶病毒致癌基因,慢性轉化的病毒則需要更長的時間引起癌症。
找出癌症最初發生的原因是不可能的。然而在分子生物學技術幫助之下,找出腫瘤內基因的異常則是可行的。因此根據基因與染色體變化的嚴重程度,對於預測病患预后情形上有迅速的進展。例如有些帶有瑕疵p53基因的腫瘤細胞,在進行化學治療時較不會發生細胞凋亡,可以預知這樣的病患會有較差的預後。基因發生突變後,細胞重新產生正常細胞沒有的端粒酶則能去除細胞分裂次數的障礙,使細胞能無限的生長分裂[16],有些突變則能使腫瘤細胞進行惡性轉移到身體其他部位,或是促進血管新生讓腫瘤細胞能得到更多營養的供應。
惡性腫瘤細胞有以下幾個特性[25]:
- 不受細胞凋亡機制的影響
- 不受限制的生長(不死,由於有大量端粒酶,細胞不受到細胞衰老機制的調控)
- 自給自足的生長因子
- 對於限制生長因子的控制不敏感
- 細胞分裂速率加快
- 重新獲得分化能力
- 不受細胞間接触抑制所影響
- 具有侵入周邊組織的能力
- 進行遠端轉移到其他部位
- 能促使血管新生[26]
細胞變化成為腫瘤細胞時,並不是一次就具有惡性腫瘤細胞的所有性質,而是借着一代一代的傳遞獲得及篩選後,變化成為最無法控制的惡性腫瘤細胞。此過程稱為複製細胞的演化,在最初僅有一些DNA發生改變,通常是產生點突變,使得細胞的基因變的不穩定。對於細胞的生長可能會產生某些影響,而經由一代又一代的細胞分裂之後,原本已帶有的突變可能會因為環境因子等刺激產生的新突變共同作用,這樣的基因體的不穩定可能由點突變增加到失去整條染色體或者發生染色體重複,所以仔細觀察癌細胞可發現通常染色體的數目並非正常的23對[5]。一個細胞要轉變並持續生長成為腫瘤並不是一件容易的事,僅僅是不受控制的生長與複製,並不能造成身體的嚴重影響。所以能否具有能力引起血管新生和轉移就成為癌細胞是否能持續生長的重要條件。對於不停複製生長的細胞來說,養分供應是極重要的。也因此才有良性腫瘤與惡性腫瘤之分別。組成良性腫瘤的細胞,可能已經具有持續生長與不受細胞間接觸抑制調控的能力,所以才能夠成為一團細胞。但也受限於細胞本身並沒有獲得轉移的能力,所以才會待在原處,和身體和平共存。DNA甲基化的模式改變也會活化或抑制基因的表現。上皮細胞等常常需要進行細胞分裂的細胞,的確是比不常分裂的細胞,例如神經元,有較高的機率轉變成腫瘤細胞。
参考资料
- (英文)Wennerberg K, Rossman KL, Der CJ. . J. Cell. Sci.. 2005, 118: 843–6. PMID 15731001. doi:10.1242/jcs.01660.
- (英文)Fearon ER, Vogelstein B. . 《细胞》. 1990, 61 (5): 759–67. PMID 2188735. doi:10.1016/0092-8674(90)90186-I.
- (英文)Fodde R, Smits R. . 《科学》. 2002, 298: 761–3. PMID 12399571. doi:10.1126/science.1077707 (英语).
- (英文)Goyette MC, Cho K, Fasching CL, Levy DB, Kinzler KW, Paraskeva C, Vogelstein B, Stanbridge EJ. . Mol. Cell. Biol.. 1992, 12: 1387–1395. PMID 1347643 (英语).
- Halazonetis TD, Gorgoulis VG, Bartek J. . Science: 1352–5, 2008 PMID 18323444. doi:10.1126/science.1140735.
- Sherr CJ. . Cell: 235–46, 2004 PMID 14744434. [2017-11-28]. (原始内容存档于2018-06-11).
- Meulmeester E, Jochemsen AG. . Curr Cancer Drug Targets: 87–97, 2008 PMID 18336191.
- Wang W, El-Deiry WS. . Curr Opin Oncol: 90–6, 2008 PMID 18043262. [2017-11-28]. doi:10.1097/CCO.0b013e3282f31d6f. (原始内容存档于2013-05-18).
- Fingerman IM, Briggs SD. . Cell: 690, 2004 PMID 15186770. [2017-11-28]. doi:10.1016/j.cell.2004.05.021. (原始内容存档于2018-06-25).
- Joerger AC, Fersht AR. . Oncogene: 2226–42, 2007 PMID 17401432. doi:10.1038/sj.onc.1210291.
- Attardi LD, Donehower LA. (05)00152-1 请检查
|url=值 (帮助). Mutat. Res.: 4–21, 2005 PMID 16038709. [2017-11-28]. doi:10.1016/j.mrfmmm.2004.08.022. (原始内容存档于2021-12-24). - Bowman T, Symonds H, Gu L, Yin C, Oren M, Van Dyke T. . Genes Dev.: 826–35, 1996 PMID 8846919.
- Fazeli A, Steen RG, Dickinson SL; et al. . Proc. Natl. Acad. Sci. U.S.A.: 10199–204, 1997 PMID 9294187.
- Matoba S, Kang J, Patino W, Wragg A, Boehm M, Gavrilova O, Hurley P, Bunz F, Hwang P (2006). "p53 regulates mitochondrial respiration." Science 312 (5780): 1650-3. PMID 16728594
- Fodde R, Smits R (2002). "Cancer biology. A matter of dosage." Science 298 (5594): 761-3. PMID 12399571
- Hinds PW, Weinberg RA. . Curr. Opin. Genet. Dev.: 135–41, 1994 PMID 8193533.
- Connolly MJ, Payne RH, Johnson G; et al. . Hum. Genet.: 122–4, 1983 PMID 6654325.
- Benedict WF, Murphree AL, Banerjee A, Spina CA, Sparkes MC, Sparkes RS. . Science: 973–5, 1983 PMID 6336308.
- Mantovani A, Allavena P, Sica A, Balkwill F. . Nature: 436–44, 2008. PMID 18650914. doi:10.1038/nature07205.
- Narisawa-Saito M, Kiyono T. [onlinelibrary.wiley.com/doi/10.1111/j.1349-7006.2007.00546.x ] 请检查
|url=值 (帮助). Cancer Sci.: 1505–11, 2007 PMID 17645777. doi:10.1111/j.1349-7006.2007.00546.x. - Hausen H (1991). "Viruses in human cancers." Science 254 (5035).
- Hanahan D, Weinberg RA. (00)81683-9 请检查
|url=值 (帮助). Cell: 57–70, 2000 PMID 10647931. [2012-04-16]. (原始内容存档于2021-12-24). - Folkman J. . Symp Soc Dev Biol: 43–52, 1974 PMID 4600889.
- (英文)Hanahan D, Weinberg RA. . 《细胞》. 2011, 144: 646-74 [2012-04-16]. PMID 21376230. (原始内容存档于2021-04-20) (英语).