布南色林
布南色林(Blonanserin),商品名洛珊(Lonasen),[2]是新型非典型抗精神病药物(2008年1月获得PMDA批准),[3]用于治疗精神分裂症。[4]与许多其他抗精神病药物相比,布南色林具有更好的耐受性,少有錐體外症候群之症状、过度镇静与低血压等副作用。[5]与许多第二代非典型抗精神病药相比,在治疗精神分裂症阴性症状方面的疗效明显优于氟哌啶醇等第一代典型抗精神病药。[6]
 | |
 | |
| 臨床資料 | |
|---|---|
| 商品名 | 洛珊(Lonasen) |
| 给药途径 | 口服 |
| ATC碼 |
|
| 法律規範狀態 | |
| 法律規範 |
|
| 藥物動力學數據 | |
| 生物利用度 | 55%[1] |
| 药物代谢 | CYP3A4[1] |
| 生物半衰期 | 12小时[1] |
| 排泄途徑 | 59%(尿液),30%(粪便)[1] |
| 识别 | |
| |
| CAS号 | 132810-10-7 |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.211.656 |
| 化学 | |
| 化学式 | C23H30FN3 |
| 摩尔质量 | 367.51 g·mol−1 |
| 3D模型(JSmol) | |
| |
医疗用途
在日本与韩国,布南色林被用于治疗精神分裂症。但在美国并没有被采用于治疗。[7]
不良反应
同于其他许多非典型抗精神病药物,布南色林会引发心血管代谢风险。虽然布南色林的副作用,如体重增加、胆固醇与三酸甘油酯指数、血糖指数和其他血脂指数之变化,与其他非典型抗精神病药物并无太大区别。但布南色林的副作用较轻,尤其是在体重增加较少的方面。[6]布南色林或可引发錐體外症候群, 以静坐不能和帕金森综合征常见。[8]
药理学
布南色林是5-HT2A受体(Ki=0.812 nM)和多巴胺D2受体(Ki=0.142 nM)的混合拮抗剂,对肾上腺素α1受体(Ki=26.7 nM)也有一定的拮抗作用。[9][10]对多巴胺D3受体有明显的亲和力(Ki=0.494 nM),[11]但对许多其他位点缺乏显著的亲和力,如5-HT1A、5-HT3、D1、肾上腺素α2、β-肾上腺素、组胺H1、蕈毒碱M1与单胺转运蛋白,[10]对Sigma受体的亲和力较低(IC50=286 nM)。[10]
对5-HT6受体具有较高的亲和力,[9][12]或许也是其最近在治疗精神分裂症认知症状方面取得较好成效的原因。其之疗效部分归功于其化学结构,与其他非典型抗精神病药物的化学结构不同,是为环辛吡啶骨架的化合物。[13]具体而言,在其独特的八分子环上添加羟基后,该化合物的(R)立体异构体对指定靶标的亲和力增强。[14]
| 受体 | Ki [nM](Blonanserin)* [9] | Ki [nM](N-deethylblonanserin)* [4] |
|---|---|---|
| D1 | 1070 | 1020 |
| D2 | 0.142 | 1.38 |
| D3 | 0.494 | 0.23 |
| D4 | 150 | - |
| D5 | 2600 | - |
| 5-HT1A | 804 | - |
| 5-HT2A | 0.812 | 1.28 |
| 5-HT2C | 26.4 | 4.50 |
| 5-HT6 | 11.7 | 5.03 |
| 5-HT7 | 183 | - |
| α1 | 26.7 (Rat brain) | 206 (Rat receptor) |
| α2 | 530 (Rat cloned) | - |
| M1 | 100 | - |
| H1 | 765 | - |
*除非另加说明,否则以下默认是为对人类受体之结果
于多巴胺D3受体上的作用
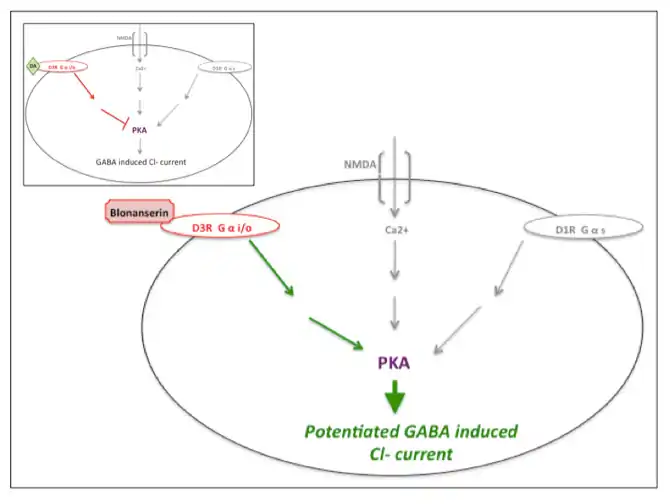 |
| 布南色林对多巴胺D3受体的作用图示,逆转了对PKA的活性(同时也受来自多巴胺D1和NMDA活性调节)的抑制,从而增强了GABA诱导的Cl-电流。插图显示了多巴胺D3受体上不间断的多巴胺活性。本图制作受启发于Hida et al. (2014) 和 Yokota 等人(2002)[11][15] |
布南色林对多巴胺D3受体起拮抗作用,可增强蛋白激酶A(PKA)的磷酸化水平,抵消多巴胺D1或NMDA受体活性的降低,从而增强GABA诱导的Cl-电流。[11][15]奥氮平似乎影响PKA的活性。[11][16]许多抗精神病药物,如氟哌啶醇、氯丙嗪、利培酮和奥氮平,主要拮抗5-HT2A和多巴胺D2受体,然而对多巴胺D2或3受体作用未知。[11][13]
药代动力学
对于BMI于19–24 kg/m2之间、体重大于或等于50 kg的成年男性,口服剂量为4 mg,一天两次或8 mg,一天一次。[17]药物的吸收采用两室模式,两室即中心室和外周室,吸收和消除均为一阶式。[18]半衰期取决于剂量。单剂量4 mg的半衰期为7.7 ± 4.63小时,单剂量8 mg的半衰期为11.9 ± 4.3小时。[17]半衰期随剂量增而增,或因为于较低单剂量下,有更多的单个时间点浓度低于定量所需的下限。[17]
布南色林不是带电化合物,化学极性很小,表面积为19.7 Å。[19]通常认为一种能通过血脑屏障的化合物的极性表面积必须小于90 Å,或为布南色林渗透性好只因,脑浆/血浆比值(brain/ plasma ratio)高达3.88便为一证。[20]
由于布南色林具有良好的渗透性,因而其在中枢神经系统中的分布容积大于外周(中枢容积=9500 L,外周容积=8650 L),但在中枢区吸收较慢。[18]也不符合里賓斯基五規則中的标准。[19]
食物摄入量的影响
食物的摄入会减缓布南色林的吸收速率,并增加外周生物利用度,而非中枢生物利用度。[18]服单次空腹剂量是安全的,进食量对其的影响与否,或是布南色林与肠道中细胞色素P450 3A4的相互作用所致。[17]
另见
参考文献
- Wen YG, Shang DW, Xie HZ, Wang XP, Ni XJ, Zhang M, et al. . Human Psychopharmacology. March 2013, 28 (2): 134–141. PMID 23417765. S2CID 12623938. doi:10.1002/hup.2290.
- . news.bioon.com. [2023-10-04]. (原始内容存档于2023-10-06).
- (PDF). Tokyo, Japan: Pharmaceuticals and Medical Devices Agency. [2013-08-16]. (原始内容 (PDF)存档于2013-01-19).
- Deeks ED, Keating GM. . CNS Drugs. January 2010, 24 (1): 65–84. PMID 20030420. doi:10.2165/11202620-000000000-00000.
- Heading CE. . IDrugs. November 1998, 1 (7): 813–817. PMID 18465651.
- Kishi T, Matsuda Y, Nakamura H, Iwata N. . Journal of Psychiatric Research. February 2013, 47 (2): 149–154. PMID 23131856. doi:10.1016/j.jpsychires.2012.10.011.
- Wang SM, Han C, Lee SJ, Patkar AA, Masand PS, Pae CU. . Clinical Neuropharmacology. 2013, 36 (6): 223–238. PMID 24201235. S2CID 21426260. doi:10.1097/wnf.0b013e3182aa38c4.
- . : p.80.
- Tenjin T, Miyamoto S, Ninomiya Y, Kitajima R, Ogino S, Miyake N, Yamaguchi N. . Neuropsychiatric Disease and Treatment. 2013, 9: 587–594. PMC 3677929
 . PMID 23766647. doi:10.2147/NDT.S34433 .
. PMID 23766647. doi:10.2147/NDT.S34433 . - Oka M, Noda Y, Ochi Y, Furukawa K, Une T, Kurumiya S, et al. . The Journal of Pharmacology and Experimental Therapeutics. January 1993, 264 (1): 158–165. PMID 8093723.
- Hida H, Mouri A, Mori K, Matsumoto Y, Seki T, Taniguchi M, et al. . Neuropsychopharmacology. February 2015, 40 (3): 601–613. PMC 4289947 . PMID 25120077. doi:10.1038/npp.2014.207.
- Tenjin T, Miyamoto S, Miyake N, Ogino S, Kitajima R, Ojima K, et al. . Human Psychopharmacology. January 2012, 27 (1): 90–100. PMID 22278973. S2CID 205925034. doi:10.1002/hup.1276.
- Suzuki K, Hiyama Y, Une T, Fujiwara I. . Analytical Sciences. November 2002, 18 (11): 1289–1290. PMID 12458724. doi:10.2116/analsci.18.1289 .
- Ochi T, Sakamoto M, Minamida A, Suzuki K, Ueda T, Une T, et al. . Bioorganic & Medicinal Chemistry Letters. February 2005, 15 (4): 1055–1059. PMID 15686911. doi:10.1016/j.bmcl.2004.12.028.
- Yokota K, Tatebayashi H, Matsuo T, Shoge T, Motomura H, Matsuno T, et al. . British Journal of Pharmacology. March 2002, 135 (6): 1547–1555. PMC 1573270 . PMID 11906969. doi:10.1038/sj.bjp.0704608.
- Nagai T, Noda Y, Une T, Furukawa K, Furukawa H, Kan QM, Nabeshima T. . NeuroReport. February 2003, 14 (2): 269–272. PMID 12598744. S2CID 41717348. doi:10.1097/00001756-200302100-00023.
- Chen X, Wang H, Jiang J, Chen R, Zhou Y, Zhong W, et al. . Clinical Drug Investigation. March 2014, 34 (3): 213–222. PMID 24399453. S2CID 35831132. doi:10.1007/s40261-013-0167-9.
- Wen YG, Shang DW, Xie HZ, Wang XP, Ni XJ, Zhang M, et al. . Human Psychopharmacology. March 2013, 28 (2): 134–141. PMID 23417765. S2CID 12623938. doi:10.1002/hup.2290.
- . PubMed. U.S. National Library of Medicine. [2023-10-05]. (原始内容存档于2023-05-02).
- Tateno A, Arakawa R, Okumura M, Fukuta H, Honjo K, Ishihara K, et al. . Journal of Clinical Psychopharmacology. April 2013, 33 (2): 162–169. PMID 23422369. S2CID 33775568. doi:10.1097/jcp.0b013e3182825bce.